I am really excited to be at PNNL – there has been a queue of people wanting to talk about semantic data. We go over the fundamentals – if there are N ontologists in a room there are > N^2 fights. Ontologies are very close to people’s souls – they have developed their world models over years and they are similar but slightly different from everyone else’s. So we agree to keep things simple – and that’s feasible in down-to-earth physical science.
We are starting with two areas where data needs to be captured and searched (NMR spectra and Compchem). Here’s compchem
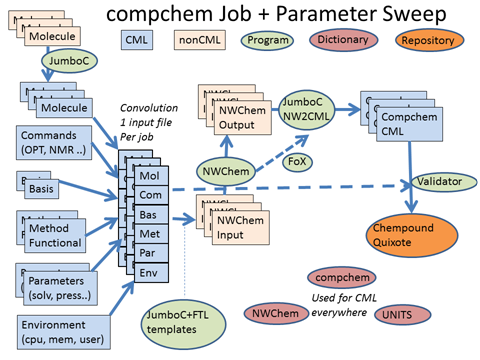
We’re using NWChem – IMO the world’s leading Open source computational chemistry (QC) code and arguably the leading one anyway. (It’s difficult to tell because the closed source ones don’t all allow benchmarks.) So NWChem is the centre. Traditionally it takes input (pink) and produces output (pink) , the point being that this is often done by hand. Some codes have GUI stuff and there’s also quite a lot of contributions from the Blue Obelisk. But what you are looking at is a leap beyond that – Quixote, dictionaries and Chempound. All, of course, Open. (Which makes it easy for any others who wish to interoperate rather than compete).
The problem with hand created input is it doesn’t scale, is desperately tedious, there are frequent human errors, little machine validation of quality, etc. We can fix all that by using Chemical Markup Language (blue). The job itself is described as a product of about 6 axes:
- Molecule. (this can also mean crystal or other extended phase). There are many legacy formats, and we can convert them all into CML
- Commands. There are a relatively common set of things we want to do. Optimisation, calculation of properties, etc.
- Basis sets. PNNL is the world leader as it produces the popular and valuable Open Basis Set Exchange BSE. Basis sets describe the atomic orbitals that we shall use to create the molecular orbitals. (don’t switch off, it gets easier)
- Methods and functionals. In simple terms how we set up Schroedinger’s equation to calculate the energy of the molecule.
- Physical Parameters. What pressure, temperature, electric field etc. to we wish to impose?
- “environment”. The local stuff about who ran the job, when, how and on what machine
Generally we can mix and match any of these (and that’s called a parameter sweep. We might have a fixed protocol and wish to apply it to 10,000 molecules. Or we might wish to take just a few molecules and try 50 basis sets (to see which gives the best agreement with experimental measurements. Or to step through a series of pressures, maybe modelling the earth’s crust.
The parameters are abstractions and independent of the program syntax. In principle they can be used with any of the main programs. The framework means that chemists don’t have to worry about the syntax. So Quixote will generate all the CML input files. These are then converted by templates (automatically) to program input (in the future we’ll use CML directly).
The program runs and creates outputs. These are normally created for human viewing but we’ve written JUMBOParser that reads them and extracts all the information into CML. This is then ingested into a Chempound repository (which holds the native input and output, the CML and also transformed RDF). And today we got the Chempound repository working here. It now ingests NWChem files. The repository is designed to allow very sophisticated searches (using RDF, SPARQL and InChI/CML). Currently very few computational chemists store their output in searchable form. Quixote is changing that.
We can be even more professional. Rather than transform and parse, we can read CML directly into the program. It needs a bit of tweaking, but that is done with FoX. FoX was written by Toby White and now tended by Andrew Walker at Bristol. FoX is a library which reads and writes XML in FORTRAN. A bit creaky compared with other languages but it works.
Then there are the dictionaries. EVERY CML component in the file must be described by a dictionary entry. That means we have to write them, and we are doing exactly that. Hard work, but necessary. Of course you don’t get impact factors for doing it, though you should.
So one more three-quarter day. I haven’t been here long enough but the momentum is unstoppable.
Do you plan, in the validation step, to include some sort of check whether the calculation is sound? I mean, if the self-consistent field iterations converged, etc.
There are two aspects:
* validate the CML against the convention (make sure all the bits are there). This is supported already
* validate against the science. This is similar to checkcif for crystallography. It will need the community to erite the rules. It will be immensely valuable. I think it would start with simple aspects (did the job declare it failed to converge?) and move on to more subjective ones later.
Yes, that’s a reasonable separation. So I suppose it would make sense to have two separate validators for that. You know, these are the kind of things people have asked of cclib, but it’s way beyond what that library was created for.
Be happy to discuss this communally.
Yes, it’s beyond cclib because you need a formal exposed semantic model. CML compchem provides some of that. You probably need validation rules in declarative format. Then it’s easier for the community to modify them
Pingback: Unilever Centre for Molecular Informatics, Cambridge - Kitware: Liberation Software « petermr's blog
Pingback: Unilever Centre for Molecular Informatics, Cambridge - #animalgarden Bottom-up Ontologies in Physical Science « petermr's blog