Antony Williams of Chemspider has offered to participate in our Open Notebook NMR experiment. Now this offer has been joined by ACDLabs – I am not sure of the formal relation between the companies but they have clear common interests. I had originally thought this was one individual making a personal offer – now there is a company that is requesting our data for them to work on.I have some genuine concerns about how we should proceed so am clearing my thoughts online. Readers will recall that I have been strongly critical of companies or nonprofits which use closed source and protect closed data. I have roughly equal numbers of correspondents who think I have been too hard on these organizations and those who think I need to be tougher. So I am taking a measured tone here.
Peter – FYI ACD/Labs are ready to participate in the work as discussed: http://www.chemspider.com/blog/?p=213#comment-3735
Peter and Tony,I think this is a fantastic project and am very keen to see how accurate the QM techniques prove to be for the subset of structures that you choose from the NMRShiftDB, and then how helpful they can be in improving the accuracy of experimental shifts in this wonderful resource.For the purposes of this work, we would be willing to provide the chemical shift predictions from the ACD/Labs software if you would like to use them in your comparison. If, for instance, they prove to be accurate enough to find many of these problems without the need for time consuming QM calculations, it may be preferrable to use the faster calculation algorithms that are available in our software. It may turn out that the ACD/Labs predictions could serve as a pre-filter to define which structures need the QM calculations and which don’t. Many variations on this theme come to mind, but we won’t know which are useful until we do the work.
Sincerely,
Brent Lefebvre
NMR Product Manager
Advanced Chemistry Development, Inc.
and Antony also requests the data:
Peter, all that is needed to perform the calculations for comparison using the ACD/Labs NMR predictors is a download of the exact dataset Christoph provided to you (we have already had issues with comparing algorithm to algorithm but using different versions of the NMRShiftDB database…not good). Also, if Nick can send us the ID of the structure inside the NMRShiftDB this should be enough. Thanks
and he points to how he/ACDLabs can predict chemical shifts:
Also, plots of the nature shown at the following page http://www.acdlabs.com/products/spec_lab/predict_nmr/chemnmr/ are generally of value when comparing improvements in algorithm performance. it’s a good way to compare your own improvements in algorithm (or Henry’s) as well as between algorithms
The link here points to a report of NMR calculations done with ACDLabs software and ChemNMR (Cambridgesoft). Note that both companies have developed their own software for chemical predictions, but the software is closed in that it can only be used if purchased, and anyway the algorithms are not visible. Note that Cambridgesoft patent algorithms – I don’t know whether ACDLabs do. The data are also closed in that they are not generally available and I have no idea of their general chemical makeup. From the page about calculating chemical shifts
During the history of the development of the ACD/Labs NMR prediction tools we have compiled and checked data from thousands of literature articles to build databases of over 165,000 assigned structures for 1H and 13C, 8000 for 15N, 13,800 for 19F, and 22,600 for 31P. With these assignments and structures as a basis and using our correlation algorithms developed over the past ten years, it should be obvious that our fragment-based predictions will offer superior performance to any rules-based systems.Fragment-based prediction offers the opportunity to cover wide ranges of structural diversity not available to rules-based systems. It is this type of performance that has enabled Advanced Chemistry Development to become the industry leader in NMR prediction today. Rigorous testing of our capabilities relative to other software packages has resulted in our software becoming the package of choice on a worldwide level for companies such as Pfizer, Astra Zeneca, GlaxoSmithKline, 3M, Eastman Kodak Co., Millennium Pharmaceuticals and many others.
and the results:
13C NMR Prediction Comparison
|
r |
r2
|
Standard Error (ppm) |
Predicted 13C Chemical Shifts |
| ACD/CNMR 8.0 |
0.999 |
0.998 |
2.33 |
68,129 |
| CambridgeSoft ChemDraw 8.0 |
0.995 |
0.990 |
5.30 |
67,841 |
Table 2: Statisics comparing the accuracy of ACD/CNMR predictions to Cambridge Soft
Once again the table illustrates the remarkable accuracy advantage of ACD/CNMR as its error is less than half that of ChemNMR. Fully 64% of the predictions from CNMR are within 1 ppm of the experimental shift as opposed to only 32% in ChemNMR (Figure 2).
I do not know if the work is formally published in the scientific manner but it is available from an unusual source:
Please request the complete comparison document from your account manager or email sales@acdlabs.com.
This clearly raises issues that need agreeing, so I’m asking the blogosphere for comment.
- If ACDLabs use their software to make calculations on our data what is the value? We cannot repeat the calculation and have no independent assessment of whether it is valid. I’m not accusing them of fudging the results but it is extremely easy to get good results by (even unconscious) selection of data. We have heard that formal analysis of chemoinformatics papers shows that many are suspect because of over- or under-fitting. Data sets and algorithms are never available, whereas we wish to make all our approach public.
- If we do make our approach and data public then they can be assimilated into ACDLabs software and operations. In principle I’m not too worried about that as long as the Open Data is honoured. But I am more concerned that ideas will be assimilated without credit – this is a general problem of Open Notebooks and one we all have to address. This includes Henry’s ideas as well as Nick’s. Note that many people deliberately use CC-NC licences (non-commercial) on their data. I have resisted this (the CC-NC on the blog is simply that I haven’t found out how to take it down and replace by CC-BY.
- If they take our data they are in a position to scoop us if, as they say, they have the best software in the world and much more data.
- I am worried by the high level of marketing in their scientific report. While it is perfectly reasonable for a company to promote its products and also reasonable to show that they outperform others there is a danger that the primary motivation is to use the current exercise for marketing rather than science and we would not wish to be associated with this.
These are clear issues but they cannot be solved easily. I am actually not clear what ACDLabs wishes to do – their data is a superset of ours as they have had access to NMRShiftDB like everyone else. I suspect our data will be cleaner as it is not easy to validate 150,000 + entries (although we have managed this with crystalEye). And I’m not quite clear what we get out of it – we can calculate empirical (fragment-based) shifts with Christoph’s software – at least enough for a reasonable filter.
But I welcome comments.
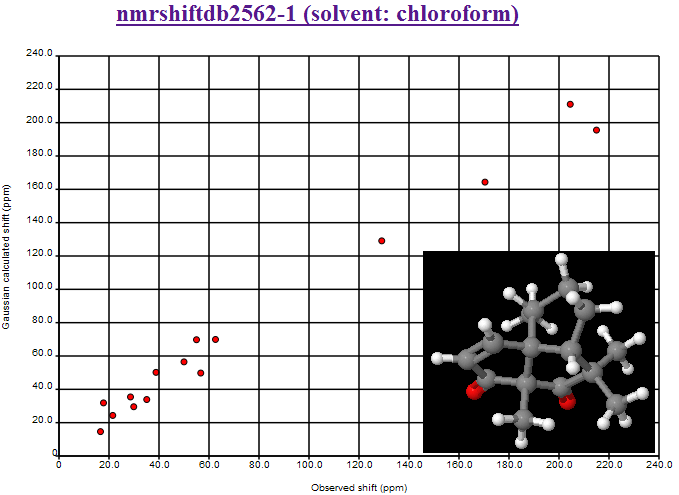
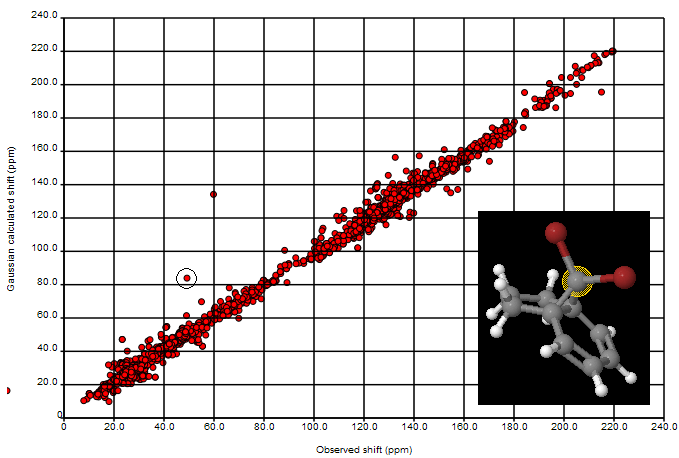 where the second largest deviation is due to the CBr2 fragment (the largest is the mistranscribed ClCN=O fragment). We shall introduce Henry’s offset’s into the calculations and re-present the data.
where the second largest deviation is due to the CBr2 fragment (the largest is the mistranscribed ClCN=O fragment). We shall introduce Henry’s offset’s into the calculations and re-present the data.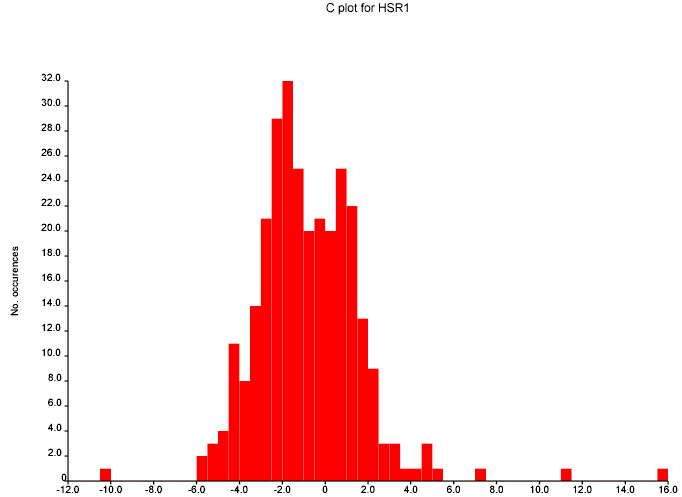


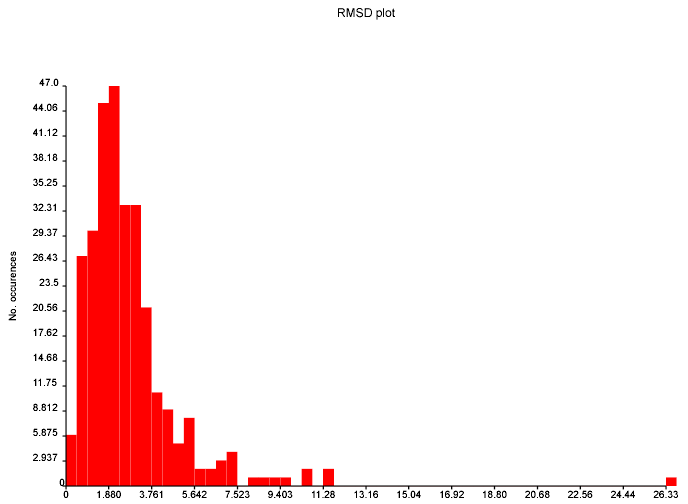
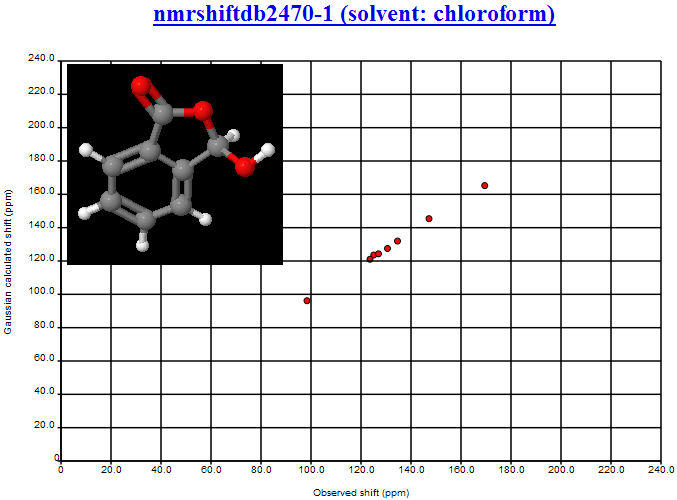
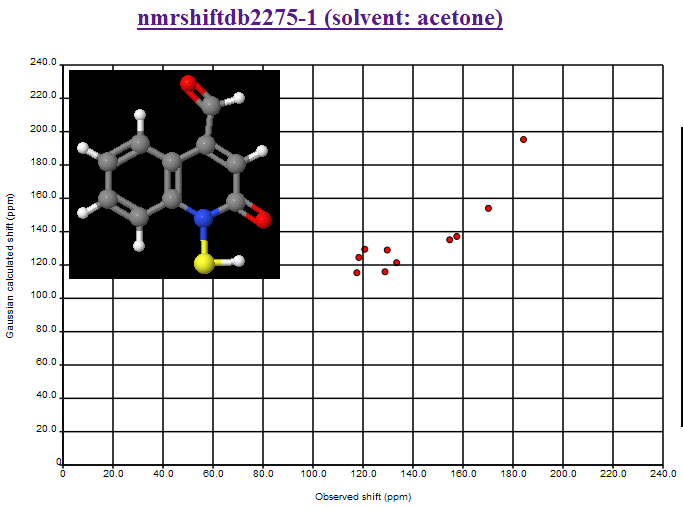
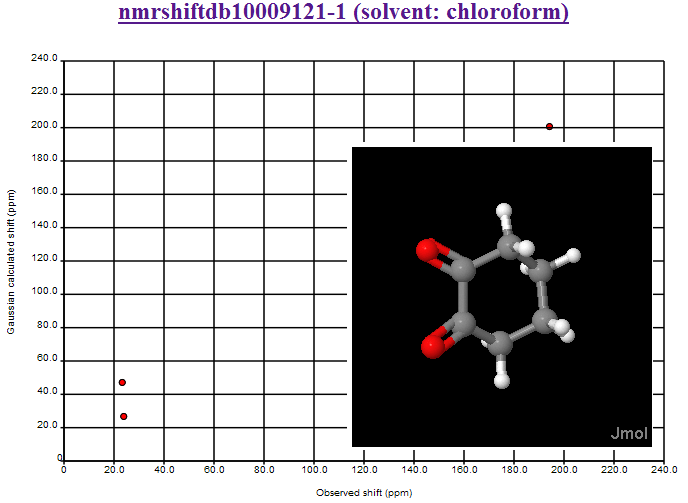
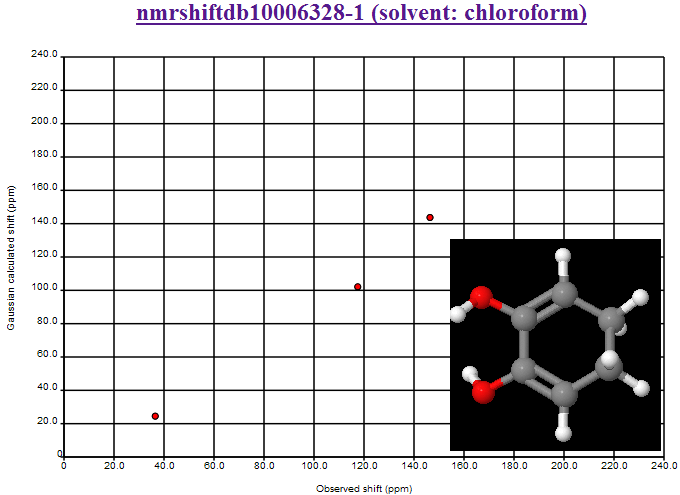
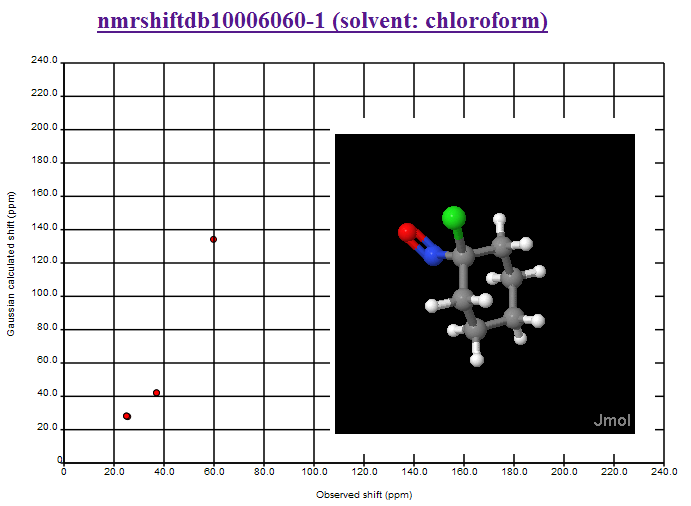
October 24th, 2007 at 1:08 pm eThe mean and variance assume a normal distribution and are sensitive to outliers. You should use the median and the inter-quartile range. This isn’t a fudge-factor – the values of the mean and variance are misleading when looking at non-normal distributions.
Also, why are you plotting the absolute value rather than the actual value? You are throwing away interesting information by folding +ve and -ve values on top of each other.
This diagram is per structure. Unless you suspect that particular structures have systematic errors, you should also do one per predicted shift. Presumably, particular environments of C atom are more difficult to calculate than others…?