There have been some general suggestions that the stereochemistry in CrystalEye structures may have errors. This is entirely possible as it’s quite difficult to check in unit tests without a large corpus of correctly annotated examples. We’d be grateful for individual examples where there seems to be errors.
At present I would like to limit this to the process of deriving atom-centered chirality (and creating a valid CML file). The relationship to SMILES and nomenclature should be decoupled at this stage. This has two steps
- deriving the CML from the CIF including 3D coordinates and hence the
<cml:atomParity>elements (these will normally be on Carbon). - deriving the wedge/hatch
<cml:BondStereo>elements. Note that these are ALWAYS arranged to have the narrow end on the stereochemical atom regardless of any false persepective. Only the local coordination of the atom matters, not the overall 2D coordinates nor the stereochemistry of atoms bonded to the current chiral centre.
I’ll leave Nick to outline the actual code steps. JUMBO is used to derive the 3D Cartesian coordinates and CDK is used to derive the 2D coordinates. It is possible to carry out the assignment of wedge and hatch bonds completely in JUMBO – I’m not sure about CDK. This post is to show how the consistency of the results can be checked. There are a number of places where errors are possible, and here we shall OMIT:
- analysis of the chirality of the overall structure derived from the anomalous scattering of the “heavy” atoms. This is measured by, e.g., the Flack parameter, (IUCr) CIF Definition data_refine_ls_abs_structure_Flack This parameter can vary between 0.0 (“correct”) and 1.0 (“wrong”) with 0.5 (“undeterminable”).
- Indications of absolute stereochemistry from the name of the compound as reported by the authors.
- Signs of torsion angles reported by authors in the CIF.
- any diagrams in the fulltext
We believe that we transform the CIFs consistently so that all structures when viewed in Jmol should have “correct chirality”. We also believe that Jmol is consistent in what it displays. We assume therefore that the coordinates and their chirality are as the author intended. Note, however, that some structures might contain both left- and right-handed molecules. Apart from the racemic case which has necessarily 50% of each this is probably uncommon, but where it happens we muct be careful in the selection of the molecule we represent.
We then calculate the atomParitys but do not normally check them by hand routinely as a higher throughput of checking can be obtained from the 2D diagrams with wedge and hatch. This relies, of course, on the 2D/wedge code being correct and this is what we need to feel confident about. Here is how it can be done fairly quickly. We take examples from the latest RSS feed of CrystalEye.
Here’s an example:

(The overlapping groups at the SE have no chirality and represent a tri-isopropylsiloxymethyl group). There are three chiral centres and the code has added H atoms to all to make the chirality clear. Here is the corresponding Jmol display (part of the Benzoyloxy, NW and silyloxymethyl SE are clipped)
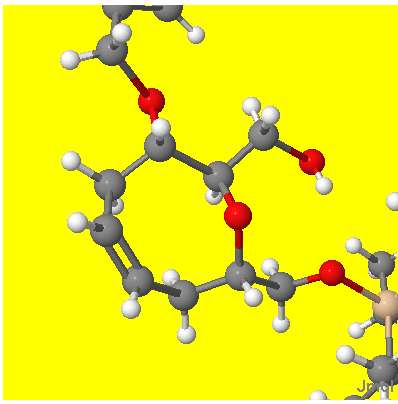
I hope you agree that the ligand atom positions round the three centres represent the same chirality as implied by the 2D picture. Remember that the convention is that the chiral atom is at the narrow end of the wedge/hatch.
If you have any cases where you believe that the 2D and 3D images do not correspond, please let us know – that’e the only point under consideration at this stage. It takes a few seconds to rotate the Jmol display till the molecule is roughly in the same orientation as the appropriate part(s) of the 2D diagram, so it’s a quick check.
[For interest only at this stage. The Silicon atom has enough scattering power that the absolute configuration can be determined with some certainty. The relevant Flack parameter is:
<scalar dictRef="iucr:_refine_ls_abs_structure_flack" dataType="xsd:double" errorValue="0.19">-0.17</scalar>
Read this as “the Flack parameter -0.17 is less than one estimated standard deviation (0.19) from 0.0. 0.0 indicates a strcuture which has been refined against the correct enantiomer”. So the crystallographi experiment asserts that it is highly likely that the chirality you see is the correct one.
The authors do not report any chemical name in the CIF (it’s possible that this can be found in the fulltext or SI) so it’s not possible to use that to confirm the stereochemistry is correct.]