Nick has more-or-less finished the computational NMR work on compounds from NMRShiftDB and we are exposing as much of the work as technically possible. Here is his interim report, some of which I trailed yesterday. The theoretical calculation (rmpw1pw91/6-31g(d,p)) involves:
- correction for spin-orbit coupling in C-Cl (-3 ppm) and C-Br (-12 ppm)
- averaging of chemically identical carbons (solves some, but not all conformational problems)
- extra basis set for C and O [below]
====== Gaussian 03 ====
–Link1–
%chk=nmrshiftdb2189-1.chk
# rmpw1pw91/6-31g(d,p) NMR scrf(cpcm,solvent=Acetone) ExtraBasis
Calculating GIAO-shifts.
0 1
C 0
SP 1 1.00
0.05 1.00000000 1.00000000
****
O 0
SP 1 1.00
0.070000 1.0000000 1.0000000
****
====== Gaussian 03 ====
In general his/our conclusions are:
- the major variance in the observed-calculated variate is due to “experimental” problems (“wrong” structures, misassignments)
- significant variance from unresolved conformers and tautomers
- small systematic effects in the offset depending on the hybridization [below]
The final variance is shown here (interactive plot at (http://wwmm.ch.cam.ac.uk/data/nmr/html/hsr1_hal_morgan/solvents-difference/index.html) requires Firefox):
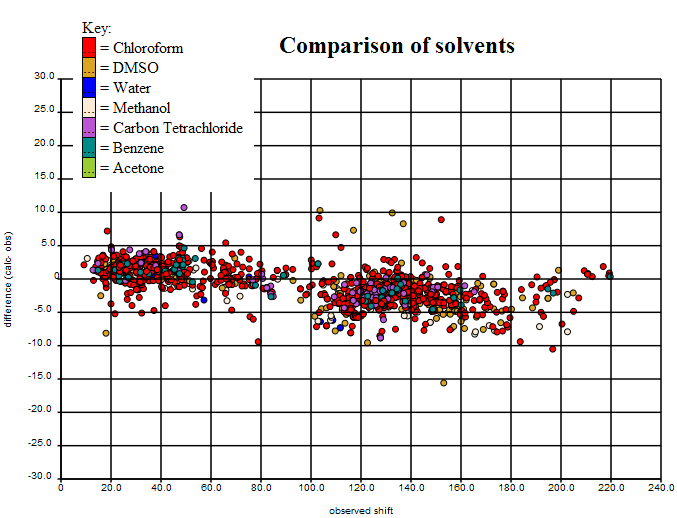
(In the interactive plot clicking on any point brings up the structure, and the various diagnostics plots can then be loadad for that structure). It can be seen that the sp3 Carbons (left) are systematically different from the sp2 (right) and we shall be playing with the basis sets to see if we can get this better. If not it will have to be an empirical calculation.
The variance can be plotted per structure in terms of absolute error (C) and intra-structure variance (RMSD). Here’s the plot (http://wwmm.ch.cam.ac.uk/data/nmr/html/hsr1_hal_morgan/RMSD-vs-C/index.html) for this (which obviously includes some of the variance from the systematic error above):
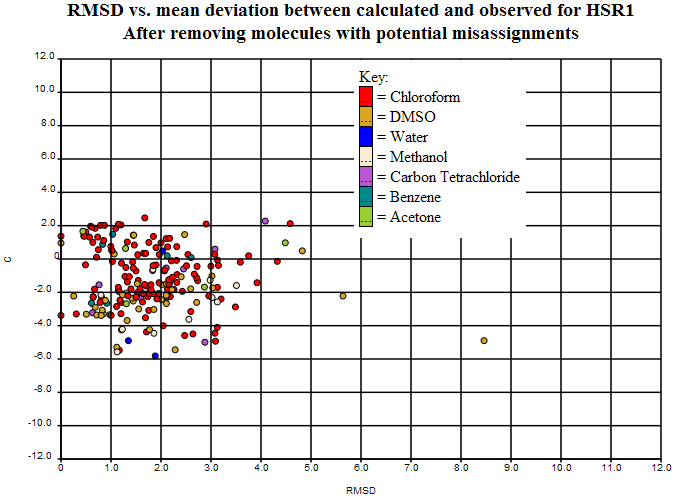
The sp2/sp3 scatter can be seen at the left but the main RMSD (> 3.0 ppm) is probably due to bad structures and unresolved chemistry. There are 22 points there and we’d be very grateful for informed comment.
Assuming the main outliers can be discarded for legitimate reasons (not just because we don’t like them) then I think we have the following conclusion. For molecules with:
- one major conformation …
- … and where there are no tautomers or we have got the major one …
- … and where the molecule contains only C, H, B, N, O, F, Si, P, S, Cl, Br …
then the error to be expected from the calculation is in the range 1-2 ppm.
We can’t go any further without having a cleaner dataset. We’d be very interested if anyone can make one Open. But we have also have some ideas how to start building one and we’d be interested in collaborators.
We’ve now essentially exposed all or methodology and data. OK, it wasn’t Open Notebook Science because there were times when we didn’t expose things. But from now on we shall try to do it as full Open Notebook Science. There may be some manual procedures in transferring results from the Condor system to web pages, but that’s no different from writing down observations in a notebook – there will be a few minutes between the experiment and the broadcast. And this will be an experiment where anyone can be involved.
Great interactive observed shift/diff(obs-calc) plot! Cheers!
(1) Nick and Joe Townsend have been responsible for all of that
Comment to: nmrshiftdb10008877 (solvent: dmso)
– wrong structure
– not 5-Methyl-2-thiouracil
– shifts correspond to 6-Methyl-2-thiouracil
– compare SDBS 4075
– compare NMRShiftDB 20027378
– structure changed to 6-Methyl-2-thiouracil
Comment to: nmrshiftdb10006008-2
– wrong assignment (C-6, C-8)
– C-6 and C-8 should be reversed (124.6 and 138.4)
– misassignment removed in NMRShiftDB
(3) This is wonderful – thanks. I knew something must be wrong but I didn’t have the knowledge to correct it.
(4) Yes, this seemed the problem but again I didn’t have the knowledge
nmrshiftdb 10006444
– stereochemistry not given (no wedge bonds)
– shifts belong to ENDO isomer (diagnostic shift 53.2 ppm)
– QM calculation used EXO isomer
– compare nmrshiftdb 10018819 (only ONE isomer, but two
different shift datasets belonging to endo and exo isomer).
nmrshiftdb 10005648
– wrong assignment (C-5, C-6)
– shifts 125.9 and 134.5 reversed
nmrshiftdb 10008573
– wrong assignment (C-6, C-8)
– shifts 119.3 and 124.1 reversed
nmrshiftdb 10006318
– many wrong assignments
– re-assigned according to my personal experience
and using common tools (ACD, CSEARCH)
nmrshiftdb 10006273
– shifts 113.9 and 116.6 reversed
(snip) …. nmrshiftdb 10006318
– many wrong assignments
– re-assigned according to my personal experience
and using common tools (ACD, CSEARCH)
…..(snip)
(1) **NOW** NMRshiftDB moves the right way – therefore my critique from MARCH 2007 was absolutely correct
(2) Interesting to see which toolset has been used: ACD & CSEARCH
(3) In the past (=since MAY 2007) the errors have been corrected, which have been found either by me or ACD ( in historical sequence) – now the errors found by QM-calculations are under correction – THEREFORE AGAIN my question: Christoph, please let me know which internal error-checks are available?
( As I stated earlier: Any database in this field has some impact on further assignments, therefore error-checking and subsequent error-correction is a matter of RESPONSIBILITY – again my question: Which protocol(s) has/have been applied to NMRShiftDB ? Answer is still missing ! Christoph, it would be nice if you could spend a few ‘*OPEN* words’ to the community with respect to this question!)
(6) Many thanks hko. Nice to see an example of sterochemistry being trapped
(7) Many thanks Wolfgang.
Minor corrections done (http://nmrshiftdb.ice.mpg.de/nmrshiftdb)
nmrshiftdb 10008719 (112.7, 118.1 reversed)
nmrshiftdb 10008864 (139.5, 142.9 reversed)
nmrshiftdb 10008906 ( 20.1, 31.8 reversed)
nmrshiftdb 10009236 ( 72.8, 78.1 reversed)
(9) Many thanks again hko. I’m guessing that most of the entries come from rather old datasets – is that right? Do you have any idea what gave rise to the errors? Human transcription? Lack of tools for assignment?
(10) Main possible error reasons:
– data come partially from very old data sources
– e.g. “blue book” (1976, Bruker 13C Data Bank)
– no prediction tools available for error checking
(9) I agree to the reassignments and structure revisions proposed by ‘hko’
(10) PMR: I’m guessing that most of the entries come from rather old datasets – is that right?
WR: That’s correct, but most of the molecules are fairly small ….. many assignments e.g. for steroids have been done around the same time and most of them were correct !
PMR: Do you have any idea what gave rise to the errors?
WR: YES – I have !
PMR: Human transcription?
WR: NO, usually not – ‘wage slaves’ do mainly a quite good job ! All the errors are coming from ONE, SINGLE source – a least 4 humans have tranfered independently the data into the computer – they did a quite good job, because the entries are CONSISTENT, but CONSISTENTLY WRONG !
PMR: Lack of tools for assignment?
WR: In most cases there were sufficient techniques available in the late ’70’s, early ’80s to do correct assignment – they were either not applied or misinterpreted …..
Within CSEARCH you can do a wonderful combination of partial structure search and line search:
Step 1: Search for fragments of the type C=C-C=O
Step 2: Select only those where the alpha-C has the larger shiftvalue than the beta-C
Step 3: Exclude functional groups which might have a strong effect like -C#N, -I, etc.
Now print a list of the literature citations and you have all the people within our community, who believe that the electronegative effect of a C=O group overrides the conjugation – believe me, it’s a long list of names !
One out of many standard protocols for data-checking within my collection …..
(11) (12) Many thanks to both of you.
It looks as if it would be useful to have a modern set of Open data for small molecules
(13) PMR: It looks as if it would be useful to have a modern set of Open data for small molecules
WR: we need ‘CORRECT’ data – many assignments of the early 70’s are absolutely correct and useful for comparison – we are suffering from the fact, that there is a collection of 20K spectra, which has never been checked for assignment errors …….
As a consequence of your QM-calculations 10 assignment corrections and 1 structure revision within a few hundred compounds have been performed by ‘hko’ (see postings above) – this corresponds to an
error rate of approx. 5% ! These errors are mainly known or can be found within a few seconds/minutes of CPU-time with HOSE/NN/etc.-methods too (I clearly state here: I appreciate your verification by QM too)
In order to show how easily such errors can be detected during daily routine-application using approx. 2 seconds of CPU-time I have choosen the ‘menthofuran-example ( NMRShiftDB: 10008906)
Details on:
http://nmrpredict.orc.univie.ac.at/csearch_summary/Dataevaluation.html
The technologies presented here and their daily application reflect MY RESPONSIBILITY for the scientific community when providing data on which other people might build their decisions …..
When asking a fully justified question, like ‘which INTERNAL data-checking protocol is/was applied to the NMRShiftDB-data’ no answer has been given here or anywhere else (Please correct me and post the corresponding URL here, if I am wrong). The basis, before discussing the OPEN/CLOSED/FREE_OF_CHARGE/PAY_PER_VIEW-question – is simply to agree on minimum standards for data-checking/data-evaluation/quality-control/etc. or more basically spoken on some guidelines for ethics in science …..
Pingback: Unilever Centre for Molecular Informatics, Cambridge - petermr’s blog » Blog Archive » Open NMR and OSCAR toolchains
Pingback: Unilever Centre for Molecular Informatics, Cambridge - Ramblings » Blog Archive » To Flash or SVG?