Egon asks about the quality of the prediction:
Egon Willighagen Says:
November 5th, 2007 at 5:58 pm e
Peter/Henry, I was wondering about the carbon shifts for atoms in an aromatic ring? Can the QM method you are using work that out? Or can it not distinguish between C=C atoms and C:C atoms (SMILES notation)? The shift differences might really be too small… if I look up C6H6 and C6H8, they are indeed small.
PMR: Nick is the expert here and I’m waiting to talk with him. However here is the first aromatic structure on my list (nmrshiftdb2470-1 (solvent: chloroform)) – I haven’t gone searching.
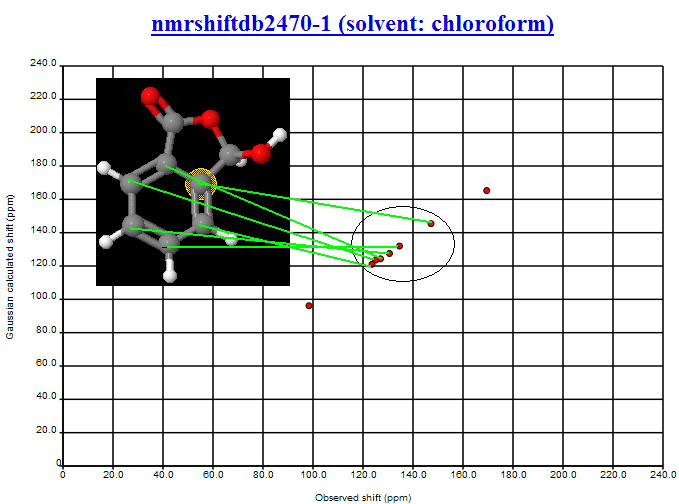
PMR: You can see the agreement between obs and calc is good. The maximum deviation seems to be about 2 ppm. The aromatic region is quite disperse (the two substituents have widely different 13C shifts so that probably helps). The NMR confirms the assignment in NMRShiftDB. I would feel reasonably happy that the shift differences were large enough to identify the atoms. Obviously if the spread was smaller it would be harder. I’m waiting for Nick to collect all the data and mount it.
Note that this is an intra-molecule comparison and I haven’t concerned myself with absolute differences. We believe there is a solvent effect on many carbonyls. Henry is better placed to comment.
EW: How about 1H NMR prediction, or can QM not do that?
PMR: Yes it can, but the dispersion is much less. 13C is from 0-200, 1H is from 0-10. So an error of 2 ppm in C is not too bad, but in 1H it’s a disaster. Also H atoms are more likely to be affected by conformation, shielding effects and solvent. So it may well be useful, but it will require a lot more work
Thanx for the elaborate answer. What I had in mind was the question whether NMR shift predictions can be used to tell me if a certain ring system is aromatic or not, and in case of fused rings, which atoms and which bonds are aromatic and which not. I’m sure the prediction error for 1H NMR shifts is well below 2ppm, and more in the order of 0.2ppm.
But maybe I should be asking, can I use CrystalEye to decide if ring systems are “aromatic”, and in case of two rings fused together (non-spiro), which atoms and bonds are aromatic and which not. Aromaticity is a fuzzy concept, with various definitions. I would be interesting in linking what the expert considers ‘aromatic’ (or SMILES, or the CDK, or …) with what the QM chemistry (via bond lengths or NMR shift predictions) and crystal structures (via bond lengths) has to teach us. The null hypothesis being that the bonds are not delocalized (bond length) and that no ring current is found (NMR shifts, 1H in particular).
Regarding those bond lengths, ‘aromatic’ bonds show a bond length in between that of single and double bonds (e.g. see [1]). The CrystalEye data does not reflect that really, and only a trimodal histograms shows up [2]. Indeed, the C#C peak is *very* low, around 1.2A 🙂 Apparently, the triple C#C bond order is underrepresented in nowadays crystallography.
Maybe aromatic C:C bonds are underrepresented too, or can the absence of a peak around 1.40A be explained otherwise? I would at least have expected a shoulder or deviation in peak shape of the peak at 1.37A.
1.http://www.chem.swin.edu.au/modules/mod2/bondlen.html (random pick)
2.http://wwmm.ch.cam.ac.uk/crystaleye/bondlengths/C-C-after-protocol.svg
(1) Henry is a world aromaticity expert so I can’t answer authoritatively there. He’s worked a lot with NMR and (I think) Nuclear Independent Chemical Shifts (NICS). Googling for this here is a typical paper:
http://www.rsc.org/publishing/journals/OB/article.asp?doi=b606070f
On the crystallography. I can’t comment on the frequencies of C-C and C:C bonds, but a lot of work has been done. I would ask Nick before trusting the C-C histogram as there are a very large number of bonds and he may have cut some out.
For each crystal that passes through CrystalEye, each unique bond (i.e. the set of bonds with no symmetry relations between them) is extracted from the crystal and added to the histograms, unless:
* one or both of the atoms are disordered
* one or both of the atoms were constrained during refinement (see _atom_site_refinement_flags in the CIF spec)
There are additional bond selection constraints for those histograms marked as ‘after-protocol’ as explained in the plot title. Apart from this, all bonds are included in the histograms.
Nick