Egon Willighagen has made a clear and appropriate statement/request of what he would like from CrystalEye:
Egon Willighagen Says:
November 5th, 2007 at 11:09 am e
Depending on the differences between the RAW and COMPLETE CMLs or maybe CIF files, I would be interested in the one or the other. I am not interested in HTML pages (TOC, indices), images, feeds, histograms, etc, as that would be something my copy would do itself.
The data corpus of CrystalEye, that’s what I would like to download. These CML files are a poor shadow of CrystalEye, only in terms of website functionality. But my interest would not be in setting up CrystalEye2, but would be to have access to the data to:
– link to CrystalEye entries (assuming the CMLs contains a CrystalEye identifier)
– derive properties myself
– test the new CDK atom typing algorithms (which chemistry is there in the CIF files that CDK cannot deal with yet)
– detect uncommon geometries
– …
PMR: This is clear and probably overlaps closely with what the crystallographic community wishes. I remind readers that this work was initiated by a summer student, Mark Holt who was sponsored by the International Union of Crystallography. Obviously, therefore, we are interested in feeding material back to the crystallographers if possible. We are also likely to be funded to continue some of the crystallographic stuff.
The information flow in CrystalEye is roughly:
- robot harvests freely accessible CIFs. Each CIF is given an address/ID based on the provenance (e.g. /publisher/journal/year/issue/DOI). A typical address is http://wwmm.ch.cam.ac.uk/crystaleye/summary//acs/joceah/2007/23/data/jo701566v/jo701566vsup1_a/jo701566vsup1_a.cif.summary.html
- For each address a page is created (“entry HTML”) which acts like a container. The RAW CML, Complete CML and CIF are addressed from this. Some publishers add copyright notices to their CIFs. While we feel this is unacceptable (because CIFs are facts), and we challenge this practice, we try to honour copyright which means that the CIFs from publishers such as ACS (above) are not held on our server.
- These CIFs are held unchanged in the entry HTML. The CIFs are then passed through CIFXML-J which converts them to a semantically identical version without added information. There should be no semantic loss and the only syntactic losses are: the precise whitespace formatting, allowed case insensitivity, ordering in the CIF and methods of quoting strings. The result is RAW-CML. If you wish to reconstruct the CIF then CIFXML-J (on sourceforge) should do this without loss. Note that RAW-CML cannot have an InChI, Cartesian coordinates, layout, bond orders, moieties, etc. I do not know whether Jmol will display it correctly (I think it may) and I am believe that Open Babel will not transform the fractional coordinates.
- RAW-CML is then fed into CIF2CML which contains a large number of transformations and heuristics to try to determine the chemical formula and other chemistry from the atom types and positions. It adds bonds, calculates moieties, iterates through that, calculates bond orders, tries to apportion formal charges, generates unique molecules (moieties) with Cartesians, calculates InChI and does a 2D layout. All this should be present in the output: CompleteCML. We expect that there may be bugs in this process due to the imprecision in creating chemistry from atom positions alone. Because CML is extensible CompleteCML should be a superset of the RAWCML – i.e. all that information is present and unchanged. But I’d welcome comments if it isn’t so.
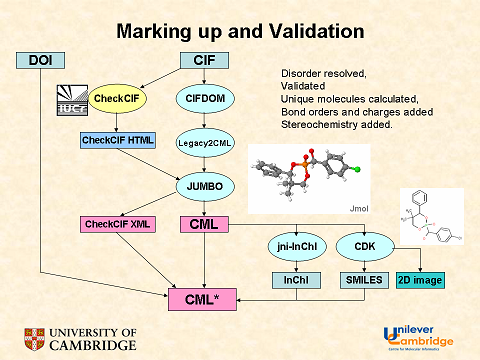
This was the process when Nick reported it at ACS. It’s changed slightly. CIFDOM is now CIFXML and it emits RAWCML. CML* represents CompleteCML. The 2D coordinates (but not the actual images) are held in CompleteCML. Complete CML also contains the moieties, each with its own InChI.
To respond to your requests:
– link to CrystalEye entries (assuming the CMLs contains a CrystalEye identifier). [PMR: The Atom feed contains the link – see above]
– derive properties myself [PMR: certainly. You should be able to work directly on the crystal structure and/or the moieties. The only thing you don’t have is the fragments.]
– test the new CDK atom typing algorithms (which chemistry is there in the CIF files that CDK cannot deal with yet) [PMR: I think there will be objective problems with coordination compounds and organometallics. We normally rely on the author to give the overall charge on a moiety. If they don’t we are usually hosed for coordination compounds.]
– detect uncommon geometries. [PMR: Certainly and we’d love to help. The bondlength plots already do this, and they are probably the first place to start. We would also plan to organise by fragments. Some fragments will have only one occurrence, others will have thousands (we would expect over 100,000 examples of phenyl groups as it can occur 10 times in some structures). Ideally we would do a cluster analysis for each fragment and then you could look for outliers – I did a brief hack at this some years back – in fact I think we corresponded. There is also enormous possibility in intermolecular interactions, perhaps a histogram of close contacts for X..Y
And we invite collaboration.
Thanks to Egon for asking his question. As I have been expressing all I want to do also is “- link to CrystalEye entries”. We will work to support the atom feeds…
“- link to CrystalEye entries (assuming the CMLs contains a CrystalEye identifier). [PMR: The Atom feed contains the link – see above]”
all that is left now is access to the backfile of entries.