Andrew Dalke raised the issue of data corruption:
PMR: Here is a widespread and almost universal example of corruption which is almost entirely down to the use of SD (MOL) files and/or SMILES in particular (but is common to almost all legacy formats). Nitric oxide (WP) is a very important molecule – it is an essential signalling molecule in the vascular system, and also a serious pollutant from transport. Its formula is NO, one nitrogen atom and one oxygen atom.
A large number of freely accessible databases give other formulas:
- eMolecules gives the formula NH=O and the molecular weight as 31.014.
- The PubChem Project gives both NH=O and the correct formula (NO)
-
Items 1 – 20 of 20
One page. 1: CID: 945 Related Structures, Literature, Other Links -
- nitrogen monoxide; nitric oxide; Nitrogen oxide …
- MW: 31.01404 | MF: HNO
2: CID: 145068 Related Structures, Assays, Literature, Other Links -
- nitric oxide; nitrogen monoxide; Nitrogen oxide …
- IUPAC: nitric oxide
- MW: 30.0061 | MF: NO
-
- Chemspider gives 4 different formulae, (HN=O, NO+, NO, NO-) all with different molecular weights.

PMR: These variations are not because there are different opinions about what “nitric oxide” is, or whether the name may be used differently by different communities. They are because the use of SD/MOL or SMILES has corrupted the information. Because SD files have no mechanism for indicating that an atom does not have implicit hydrogens, many programs are “clever” and add them according to “valence rules”. While these are OK for a subset of chemistry they are a disaster for others. Nitric oxide is just one of many examples where they fail. So that is why I cannot answer Chemspider’s request for SD files of CrystalEye – I KNOW it will corrupt the information. It is possible that there is a simple algorithm that could filter out “most” of the entries which would not be corrupted, but it will not be watertight. That is why we have developed CML – it is designed to avoid corruption.
- When a document in one format is converted to another there is likely to be information loss. Is “information loss” necessarily “corruption”? From my experience in dealing with PDB files, which has some of these crystallographic properties, I think there can be meaningful information despite the information loss. So long as the tools and the users understand that there are limitations in the conversion.
PMR: There are “obviously” parts of the information that can be omitted without corruption. An example is “iucr:_publ_contact_author_phone”. But what happens if you omit “occupancy” in an entry ? It looks like:
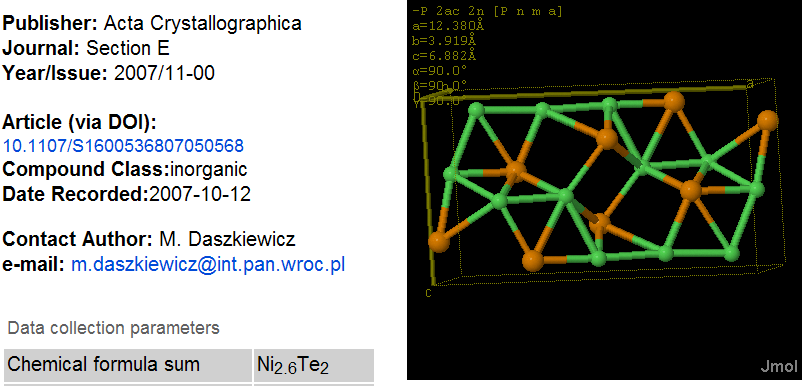
Notice that the _chemical_formula_sum contains non-integral atom counts – this is common in crystal structures nd is supported by the _atom_site_occupancy flag in CIF which points to the last field before the two dots.
_atom_site_type_symbol
_atom_site_label
_atom_site_fract_x
_atom_site_fract_y
_atom_site_fract_z
_atom_site_U_iso_or_equiv
_atom_site_adp_type
_atom_site_calc_flag
_atom_site_refinement_flags
_atom_site_occupancy
_atom_site_disorder_assembly
_atom_site_disorder_group
Ni Ni1 0.05235(10) 0.2500 0.4203(2) 0.0152(4) Uani d S 1 . .
Ni Ni2 0.14310(14) 0.2500 0.0802(3) 0.0185(4) Uani d SP 0.80 . .
Ni Ni3 0.15349(14) -0.2500 0.5956(3) 0.0191(5) Uani d SP 0.80 . .
Te Te1 0.25343(5) 0.2500 0.42149(10) 0.0131(3) Uani d S 1 . .
Te Te2 0.00373(5) 0.2500 0.78163(10) 0.0146(3) Uani d S 1 . .
Confirm that Ni(1+0.8+0.8) => Ni2.6 and Te(1+1) => Te2. CML is designed to hold this without loss (through the occupancy attribute) but SD files, SMILES and almost all other legacy (except PDB and a few other crystallographic files) are not. Therefore using SD to bundle this entry and transmit is is guaranteed to corrupt it.
[Note added later. There is a well characterised HN=O molecule – see NIST Webbook – but it is nitrosyl hydride, not nitric oxide.]
{kind=link}
November 4th, 2007 at 2:32 am e