We’ve started going through the structures in serial order. Here are two in the first 4 I looked at. One shows near perfect agreement, the other is frankly awful.
Here’ s the link to NMRShiftDB: nmrshiftdb2470-1 (solvent: chloroform)
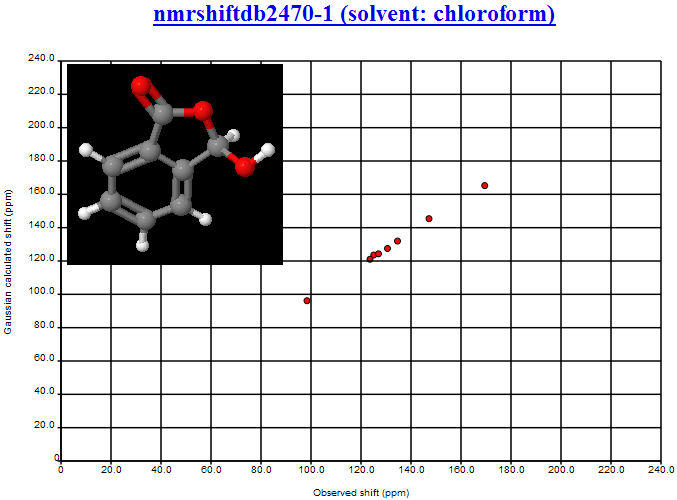
You can see that the RMS is of the order of 1 ppm (we haven’t yet calibrated the offset so it’s improper to give a clearer figure).
Now here is one that is all over the shop: nmrshiftdb2275-1 (solvent: acetone)
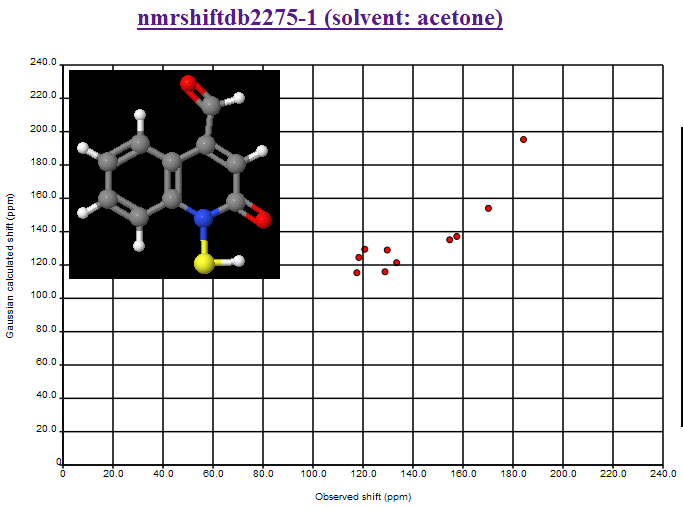
Here is the Additional data on NMRShiftDB:
|
|
Molecule (2275)
|
http://www.nmrshiftdb.org:8080/portal/_sdathe_Fri Oct 04 10:25:31 CEST 2002.002.ms
|
|---|---|
| Chemical name(s) | N-Mercapto-4-formylcarbostyril |
| Molecular weight | 205.233 |
| Number of all rings, size of smallest set of smallest rings | 3, 2 |
| CAS-Number | |
| Molecule keywords | bacteria, antibiotics, Pseudomonas fluorescens G308, |
| Type | 13C |
| Temperature [K] | 298 |
| Solvent | Acetone-D6 ((CD3)2CO) |
| Additional comments | antifungal effects in vitro against several plant pathogenic fungi; Multiplicities generated automatically from H count |
I have no idea what is going on. Nor has Christoph. So here is your chance to solve the problem. Here are some ideas I suggested
- spectrum and compound got muddled.
- compound is “wrong”. I am not clear whether N-SH groups are stable but I don’t think they are common.
- there is some serious tautomerism going on.
- this compound disobeys the laws of physics
Anyway you know as much as we do now and you have a link to the entry and the literature. Please add your comments below and if you do discover something fantastic remember where you found the data.
Copy/paste from my comment in [1]:
“The ‘bad’ is interesting… can you make a plot ‘predicted versus measured’ per solvent? That is, maybe the ‘bad’ is bad, because of the more difficult to model acetone solvent field? I remember that solvents like acetone (which is polar on one side, not so polar on the other side, where the methyls shield the dipole) are particularly difficult to model; the molecules orient in different orientations around the molecule depending on functional groups interacting with the acetone dipole…”
1.http://wwmm.ch.cam.ac.uk/blogs/murrayrust/?p=715#comments
Wouldn’t you expect the tautomer to dominate in this case with the extension of aromaticity?
(1) Good point, Egon. More later.
(2) Sounds plausible. But I am struggling to draw a tautomer. Could you make a suggestion, please (a SMILES will do).
Dear Egon;
you are absolutely correct with your remarks about solvents a few lines above !
This was exactly one of my points during my NMRShiftDB-critique: Why have 85% of the entries no solvent given in download-file ? This point went online on May 31st, 2007 and can be found on my webpage http://nmrpredict.orc.univie.ac.at/csearchlite/Robien2Ryan_May31_2007.html
BEFORE starting a project it is every scientists responsibility to look around what is already there, because the main intention is create a ‘new value’ for the community. Solvent effects were already known a few years ago ……
Comment to: NMRShiftdB Molecule (2275)
http://www.nmrshiftdb.org:8080/portal/_sdathe_Fri Oct 04 10:25:31 CEST 2002.002.ms
Chemical name(s) N-Mercapto-4-formylcarbostyril
Probably wrong structure. N-OH and C=S group would better correspond to shifts
than of N-SH and C=O group.
Assignments of some shifts are wrong.
To hko:
(1) No transcription error as already stated by PMR
(2) Using CSEARCH-NN for data-checking:
Average deviation: 9.4 ppm per carbon (horrible !)
1st correction: 8.4 ppm (as proposed by hko)
2nd correction: 5.4 ppm (reassignment of aromatic ring)
(3) Signals at 157.51 and 154.69 do not fit to this structure (both to be expected below 150)
Double bond with conjugation (either 2xC=O or 1xC=O/1xC=S) – effects should partly compensate for C10 in nmrshiftdb-numbering
(4) Have this dataset on my ‘list’ since its existence in CSEARCH about 3 years ago …… cant provide any final solution – HKO’s proposal goes the right way, but is not final (to my opinion !)
(5) Application of ANY PREDICTION SOFTWARE PACKAGE should put a lot of RED and/or YELLOW markers on this entry ….. CSEARCH does this AUTOMATICALLY during display – a NN-spectrum is calculated on-the-fly whenever you display an entry ! (ACD does it too according to the examples I have seen)
Conclusion: Guys, its all there, we only need a few drops of glue putting the pieces together …….