I posted our intermediate conclusions on Nick Day’s computational NMR project, and have received two lengthy comments. I try to answer all comments, though as Peter Suber says in his interview sometimes comments lead to discourses of indefinite length. I am taking the pragmatic view that I will mainly address comments (and subcomments) that:
- address our project as we defined it (not necessarily the project that others would like us to have done)
- add useful information (especially annotation of suspected problems)
- or show that our scientific method is flawed or could be strengthened
- relate to Open issues. In our present stage of robotic access to and re-use of data we can only realistically use databases that explicitly allow re-use of data and do not require special negotiation with the owners
There has been a great deal of discussion (far more than we had expected) on our project. Some of this has been directly relevant in responding to our direct requests for annotation of specific outliers and we acknowledge posts from Egon Willighagen, Christoph Steinbeck, Jean-Claude Bradley, Wolfgang Robien, and the University of Mainz. A lot of the discussion has been of general interest but not directly relevant to the aims of the project which was to show what fully automatic systems can do, not to create specific resources (“clean NMRShiftDB”). It is possible, though not necessary, that the work might be more generally valuable depending on what we found.
Wolfgang Robien: October 27th, 2007 at 12:03 pm e
You wrote: ….only Open collection of spectra is NMRShiftDB – open nmr database on the web.
Also the SDBS system can be downloaded – as far as I remember its limitited to 50 entries per day (should be no problem because QM-calculation are quite slow compared to HOSE/NN/Incr)
PMR: thank you for reminding us. I have not used SDBS and it looks a useful resource for checking individual structures but it is inappropriate for the current work as:
- robotic download is forbidden
- there is no obvious way of downloading sets of structures – they need to be the result of a search
- there is no obvious machine-readable connection table (there is a semantically void image).
- there are no 3D coordinates (this is not essential but it meant Nick could work almost immediately)
It is possible that if we wrote to the maintainers they would let us have a dataset, but this would double the size of the project at least.
If you need 500 entries with a certain specification (e.g. by elements, molwt, partial structure, etc.) and you want to perform a common project, please let me know …..
PMR: Thank you. This is a generous offer and we may wish to take it up. Contrary to your comment that I am an NMR expert, I’m really not – I’m an eChemist and this exercise in NMR is because I wish to liberate NMR from the pages of journals. If we find that Henry’s program needs more data or that yours has fewer problems it could be extremely valuable. We would wish the actual data to be Open so that others can re-use it.
PMR is quoted as “We downloaded the whole of NMRShiftDB. When we started we had NO idea of the quality. …”
That is quite true. There were a number of public comments on NMRShiftDB ranging from (mild) approval to (mild) disapproval, some scalar values for RMS against various prediction programs and some figures on misassignments, etc. These gave relatively little indication of the detailed data quality – e.g. the higher moments of variation.
If there is currently a full list of NMRShiftDB entries with your annotations this would be valuable. Currently I can find a number of comments on individual entries with gross problems at
http://nmrpredict.orc.univie.ac.at/csearchlite/hallofshame.html
but these seem anecdotal rather than a complete list.
PMR: and the second set of comments
ChemSpiderMan Says:October 27th, 2007 at 12:43 pm
2) Regarding “the only Open collection of spectra is NMRShiftDB – open nmr database on the web.” Just to clarify these are NOT NMR spectra actually. Unless NMRShiftDB has a capability I am aware of NMRSHiftDB is a database of molecular structures with associated assignments (and maybe in some cases just a list of shifts..maybe all don’t have to be assigned.)PMR: Thank you for the correction. I should have said peaklists with assignments.
4)Regarding “We knew in advance that certain calculations would be inappropriate. Large molecules (> 20 heavy atoms) would take too long. ” The 20 heavy atom limit is a real constraint. I judge that most pharmaceuticals in use today are over 20 atoms (xanax, sildenafil, ketoconazole, singulair for example). I would hope that members of the NMR community are watching your work as it should be of value to them but I believe 20 atoms is a severe constraint. That said I know that with more time you could do larger molecules but a day per molecule is likely enough time investment.
PMR: We have strategies for dealing with larger molecules but are not deploying them here.
6) Regarding “So we have a final list of about 300 candidates.” Out of a total of over 20000 individual structures your analysis was performed on 1.5% of the dataset. How many data points was this out of interest.
PMR: I expect about 6-20 shifts per entry. Some overlap because of symmetry
7) Regarding ” probably 20% of entries have misassignments and transcription errors. Difficult to say, but probably about 1-5%”. This suggests about 25% of shifts associated with my estimated 3000 shifts are in error. This is about 750 data points and this conclusion was made by the study of 300 molecules. For sure the 25% does not carry over to the entire database. It is of MUCH higher quality that that. My earlier posting suggested that there were about 250 BAD points. The subjective criteria are discussed here (http://www.chemspider.com/blog/?p=44). Wolfgang suggested about 300 bad points but we were both being very conservative.You discussed the difference between 250 and 300 here on your blog as you likely recall http://wwmm.ch.cam.ac.uk/blogs/murrayrust/?p=346
PMR: Nick will detail these later. We believe that the QM method is sufficiently powerful to show misassignments of a very few ppm – I will not give figures before we have down the work. With known variance it is possible to give a formal probability that peaks are misassigned. I have shown some examples of what we believe to be clear misassignments, but we have not gone back to the authors or literature (which often does not have enough information to decide). I do not believe you can compare your estimates with ours as you and we have not defined what a misassignment is.
8) Regarding “We realise that other groups have access to larger and, they claim, better data sets. But they are closed. I shall argue in a later post that closed approaches hold back the quality of scientific data.” I think your comments are regarding Wolfgang Robien and ACD/labs. That is true that we have access to larger datasets but we can limit the conversations to NMRShiftDB since we ALL have access to that. Robien’s and ACD/Labs algorithms can adequately deal with the NMRSHiftDB dataset. For the neural nets and Increment based approach over 200,000 data points can be calculated in less than 5 minutes (http://www.chemspider.com/blog/?p=213). You have access to the same dataset and can handle 300 of the structures. Your statement is moot..it is NOT about database size but about algorithmic capabilities.
PMR: My statement was about size and quality of datasets and is completely clear. It has nothing to do with algorithms. I am not interested in comparing the speed of algorithms but am concerned about metrics for the quality of data. I shan’t discuss speed of algorithms unrelated to the current project
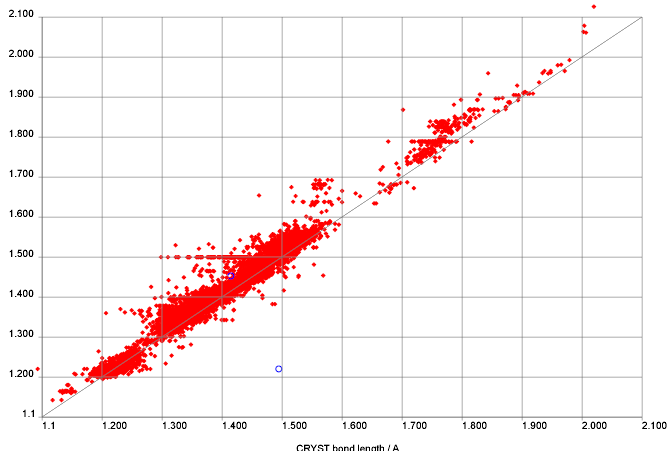
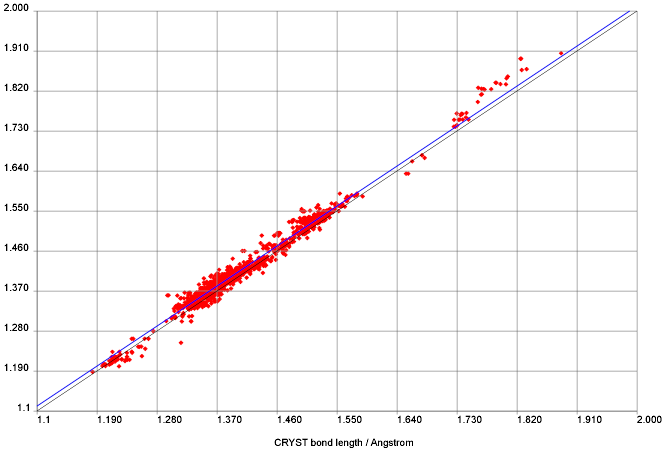

October 26th, 2007 at 1:15 am eThe first one is another misassignment. Look up the structure in the NMRShiftDB and you will see one correctly assigned and one misassigned spectrums. This kind of issues should be filed as ‘data’ bug report at:
http://sourceforge.net/tracker/?atid=560728&group_id=20485&func=browse
I’m will do this one.
October 26th, 2007 at 1:17 am eFiled as:
http://sourceforge.net/tracker/index.php?func=detail&aid=1820353&group_id=20485&atid=560728
October 26th, 2007 at 8:48 am eAnother error: Pachyclavulide-A (should be C26 instead C27), MW=510
Found automatically by the following procedure within CSEARCH:
Search all unassigned methylgroups located at a ring junction. The methylgroup must be connected either with an up or down bond. As an additional condition, it can be specified if only “Q’s” are missing or if the multiplicity of missing lines can be ignored. I think a quite sophisticated check which goes into deep details of possible error sources. […]
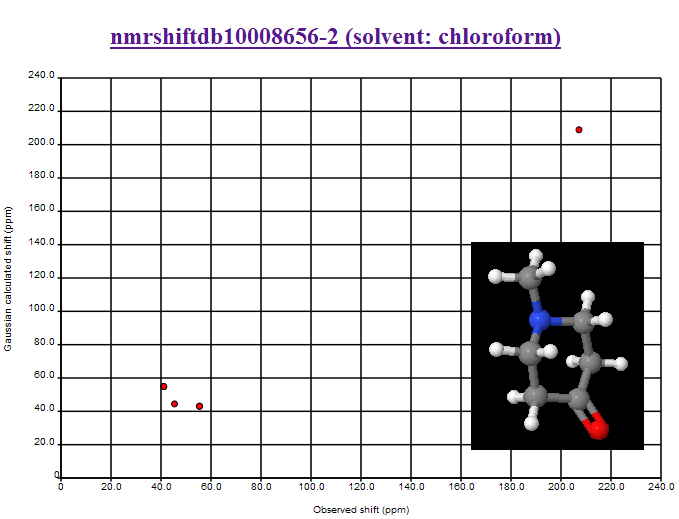
October 27th, 2007 at 12:02 pm eMisassignments NMRShiftDB (10008656-2) removed.
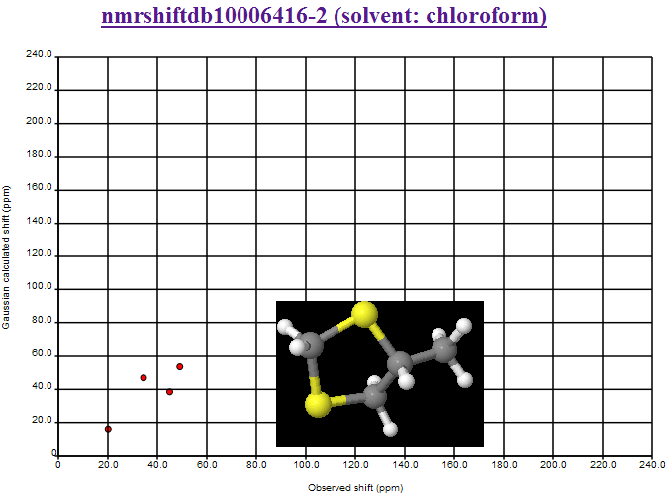
October 27th, 2007 at 5:28 pm eMisassignments NMRShiftDB (10006416-2) removed. 45.0 and 34.4 reversed.